Variabilität der pBuM189 Satelliten-DNA unter südamerikanischen Populationen von Drosophila buzzatii
Maschinenübersetzung
Der Originalartikel ist in EN Sprache (Link zum Lesen) geschrieben.
Die pBuM189 Satelliten-DNA wurde in Drosophila buzzatii Populationen analysiert, die den Großteil der Verbreitung der Art in Südamerika abdecken. Diese satDNA besteht aus A+T-reichen Monomeren von 189 bp und frühere Studien zeigten eine schnelle Rate an evolutionären Veränderungen dieses Bestandteils des D. buzzatii Genoms. Insgesamt wurden 63 pBuM189 Wiederholungseinheiten aus 14 D. buzzatii Populationen (9 aus Brasilien und 5 aus Argentinien) untersucht. Die durchschnittliche nukleotidale Variabilität unter den 63 Wiederholungen beträgt 4,2 %. Mindestens eine Wiederholung (Juan/4) scheint Teil einer anderen pBuM189 satDNA Unterfamilie zu sein. Die nukleotidale Ausrichtung aller 63 Wiederholungen zeigte keine spezifischen nukleotidalen Substitutionen oder Indels, die jede Population oder Gruppen geografisch naher Populationen unterscheiden könnten. Ein solches Fehlen von satDNA interpopulationeller Differenzierung steht im Einklang mit früheren mtDNA-Daten, die auf einen hohen Genfluss und sehr geringe Populationsdifferenzierung in der Mehrheit der D. buzzatii Verbreitung in Südamerika hinweisen. Genfluss könnte während der Vergletscherungsereignisse im Pleistozän möglich gewesen sein, wie dem, das vor 13.000 bis 18.000 Jahren stattfand, als D. buzzatii wahrscheinlich eine kontinuierlichere Verbreitung hatte als die, die heute beobachtet wird.
Die kaktusliebende Fliege Drosophila buzzatii (buzzatii-Cluster, repleta-Gruppe) hat eine weite Verbreitung in Südamerika und ist in den meisten Gebieten Argentiniens, Boliviens, Paraguays und Brasiliens anzutreffen. Die Art nutzt verrottendes Gewebe verschiedener Feigenkaktusse (Opuntia spp.) und säulenförmiger Kakteen zur Ernährung und Fortpflanzung (PEREIRA et al. 1983). In den letzten zweihundert Jahren wurden Opuntia-Kaktusse vom Menschen in mehrere Regionen der Welt transportiert, einschließlich der Länder des Mittelmeerraums, Afrikas und Australiens. Folglich wanderte D. buzzatii mit seinen Wirten. Diese Situation machte D. buzzatii zu einem geeigneten Organismus, um Fragen zu genetischen Veränderungen während des Kolonisationsprozesses zu untersuchen (FONTDEVILA et al. 1982; HALLIBURTON und BARKER 1993; ROSSI et al. 1996; FRYDENBERG et al. 2002).
Nach Daten zu chromosomalen Inversionen ist der argentinische Chaco das wahrscheinlichste Zentrum seiner Radiation (WASSERMAN 1962; CARSON und WASSERMAN 1965). D. buzzatii ist auch im Chaco sehr zahlreich, assoziiert mit einer Reihe verschiedener Kaktusarten (Vilela et al. 1980). Im Allgemeinen sind brasilianische Populationen patchy über kleine Gebiete xerophytischer Vegetation verteilt, wo sie in sehr niedrigen Frequenzen gefunden wurden (VILELA et al. 1983; TIDON-SKLORZ et al. 1994; TIDON-SKLORZ und SENE 1995). Die einzige Ausnahme ist Südbrasilien, wo relativ große Populationen gefunden wurden (VILELA et al. 1983; RUIZ et al. 2000).
Es wurde postuliert, dass D. buzzatii Brasilien durch Veränderungen in der Verteilung der ariden Vegetation während der Vergletscherungsereignisse, wie sie im späten Pleistozän auftraten, kolonisiert hat (BARKER et al. 1985; FIGUEIREDO und SENE 1992).
BAIMAI et al. (1983) analysierten die Metaphasechromosomen mehrerer D. buzzatii Populationen aus Argentinien und Brasilien. Alle untersuchten Populationen zeigen dasselbe Metaphaseblatt. BARKER et al. (1985) fanden keine signifikanten Allozyme-Unterschiede zwischen den südamerikanischen Populationen. Nur eine kleine lokale Differenzierung schien in den brasilianischen Populationen aus Bahia (Nordosten) und Tramanda´1 (Süden) stattgefunden zu haben. In einer anderen Studie berichteten FIGUEIREDO und SENE (1992), dass von 16 in Argentinien detektierten Chromosomeninversionen nur zwei in den brasilianischen Populationen vorhanden waren und keine im nordöstlichen Brasilien gefunden wurde. ROSSI et al. (1996) und DE BRITO et al. (2002) fanden hohe Ebenen der mtDNA-Einheitlichkeit unter den südamerikanischen D. buzzatii Populationen, und mehrere Tests deuteten auf einen hohen Genfluss über die meisten geografischen Verteilungen von D. buzzatii hin. DE BRITO et al. (2002) berechneten auch, dass D. buzzatii seit mindestens 100.000 Jahren in Brasilien vorhanden ist, was auf eine vorholocäne Expansion der D. buzzatii argentinischen Populationen in Richtung Brasilien hindeutet.
Um mehr Informationen über die genetische Variabilität von D. buzzatii in Südamerika zu erhalten, suchten wir nach einer nukleären DNA. Satelliten-DNA (satDNA) ist eine Klasse von hochrepetitiver nicht-kodierender DNA, die in großen homogenen Arrays im Genom nahezu aller eukaryotischen Organismen tandemartig angeordnet ist. Die normalerweise schnelle Rate des evolutionären Wandels von satDNA-Sequenzen wird durch eine Reihe von satDNA-Familien unterstützt, die sich als artspezifisch erwiesen haben (BACH-MANN et al. 1989; KING und CUMMINGS 1997).
Frühere Studien zeigten, dass satDNA-Sequenzen nützlich sein könnten, um konspezifische Populationen zu unterscheiden (ELDER und TURNER 1994; WU et al. 1999).
KUHN et al. (1999) beschrieben die pBuM189 Satelliten-DNA von D. buzzatii. Sie besteht aus Wiederholungseinheiten, die leicht AT-reich und 189 bp lang sind. Die fünf beschriebenen pBuM189-Sequenzen wurden aus der genomischen DNA nur einer D. buzzatii-Population gewonnen. Hybridisierungs- und PCR-Experimente konnten pBuM189-Sequenzen im Genom eng verwandter Arten wie D. serido, D. borborema oder D. koepferae nicht nachweisen. Daher deuten die verfügbaren Daten auf eine schnelle Rate des evolutionären Wandels dieses Bestandteils der D. buzzatii nukleären DNA hin.
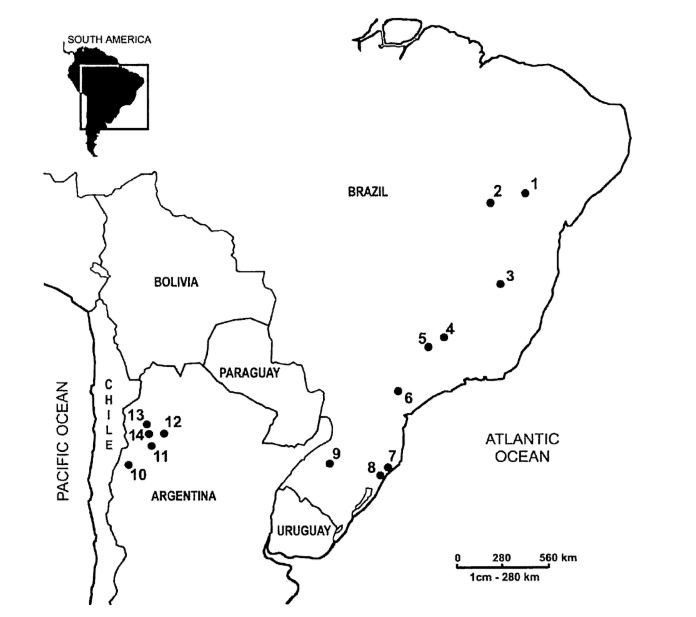
In der vorliegenden Studie analysierten wir 63 pBuM189-Sequenzen von Stämmen, die 14 D. buzzatii-Populationen (9 brasilianische und 5 argentinische) repräsentieren. Die Populationen decken den größten Teil der geografischen Verbreitung dieser Art in Südamerika ab.
Material und Methoden
Proben
Der geografische Standort aller untersuchten D. buzzatii-Populationen ist in Abb. 1 dargestellt. Die verwendeten Stämme mit ihren jeweiligen Lokalitäten sind: D69R5 (1); J79H41 (2); D54F5 (3); H86G8 (4); D42F2 (5); H99X6 (6); H42F1 (7); J25A20 (8); J28E15 (9); ArgE3 (10); Cat (11), Hondo (12); Salta (13) und Ticucho (14).
Molekulare Techniken
Die pBuM189 Sequenzen wurden ursprünglich aus dem Genom von D. buzzatii nach der DNA-Verdauung mit dem MspI-Enzym isoliert (KUHN et al. 1999). In der vorliegenden Arbeit wurde das SspI-Enzym (das ebenfalls nur einmal pro pBuM189 Wiederholungseinheit vorhanden ist) verwendet, um das Klonierungsverfahren zu erleichtern. pBuM189 satDNA Fragmente wurden durch ihr Auftreten als auffällige Bänder von ungefähr 190 bp im Hintergrundschleier der genomischen DNA identifiziert. Die DNA aus dem 190 bp DNA-Band wurde durch Übernachtinkubation in 500mM NaAc; 1mM EDTA aus dem Gel eluiert. Die zurückgewonnenen Fragmente wurden in einen pUC18 Plasmidvektor ligiert und in kompetente Escherichia coli DH5-a Zellen transformiert. Rekombinante Klone wurden als weiße Kolonien auf Ampicillin-Platten ausgewählt, die X-gal (5-Bromo-4-chloro-3-indolyl-β-D-galactosid) und IPTG (Isopropyl-β-D-thiogalactopyranosid) enthielten. Die Template-DNA-Reaktion für die Sequenzierung wurde gemäß dem Handbuch des BigDye Terminator Cycle Sequencing Ready Reaction Kit (Perkin-Elmer) vorbereitet. Die automatische DNA-Sequenzierung wurde auf einem ABI Prism™ 377 (Perkin-Elmer) Sequencer durchgeführt.
Das MEGA 2.1 Programm (KUMAR et al. 2001) wurde zur Schätzung der Nukleotidvariabilität, genetischen Distanzen und zur Konstruktion eines NJ-Dendrogramms verwendet.
Ergebnisse und Diskussion
Wie bereits aus den vorherigen pBuM189 Sequenzdaten erwartet, führte die Genom-DNA-Verdauung von 13 D. buzzatii Stämmen mit SspI zu einem stark gefärbten DNA-Band von etwa 190 bp, das den pBuM189-Monomeren entspricht, gefolgt von Bändern mit abnehmender Intensität von 380 bp (Dimere), 570 bp (Trimere) und so weiter. Das 190 bp DNA-Band aus jedem Verdau wurde unabhängig vom Gel eluiert und kloniert. Insgesamt wurden 58 Klone sequenziert, 39 aus 8 brasilianischen Populationen (Orte 1 – 4, 6 – 9) und 19 aus 5 argentinischen Populationen (Orte 10 – 14). Die Nukleotid-Ausrichtung dieser 58 Klone mit den 5 pBuM189 Wiederholungen (sert/1 – 5 in der vorliegenden Arbeit) aus dem Ort (5), der zuvor von KUHN et al. (1999) beschrieben wurde, bewies, dass sie alle pBuM189 satDNA-Monomere darstellen (Abb. 2).

Die 63 pBuM189-Wiederholungen sind leicht A+T-reich (71 %) und die durchschnittliche Sequenzvariabilität beträgt 4,2 %. Die Hauptquelle der Variation unter den 63 pBuM189-Wiederholungen sind Einzel-Nukleotid-Substitutionen, die zu 69 % wiederholungs-spezifisch sind und die verbleibenden häufig bei zwei oder mehr Wiederholungen vorkommen. Es wurden nur 11 Indels gefunden.
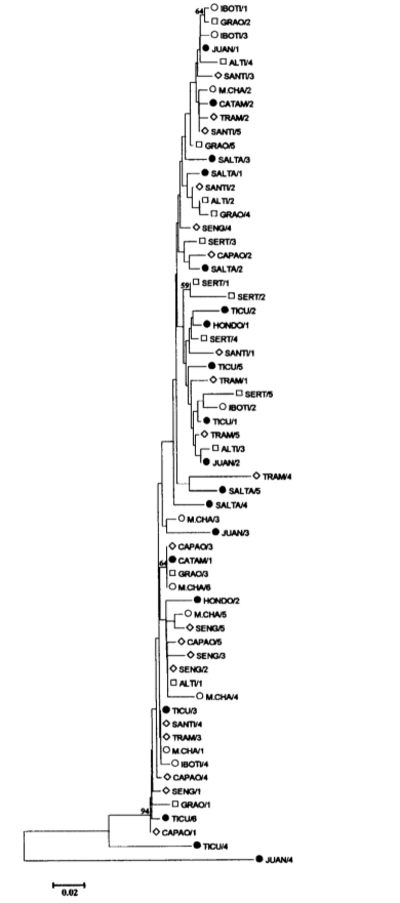
Eine pBuM189-Wiederholung scheint außergewöhnlich zu sein. Der Juan/4-Klon aus der Lokalität (10) wies in allen paarweisen Vergleichen unerwartet hohe Sequenzdivergenzen auf (im Durchschnitt 22,1 % und immer über 19 %). Darüber hinaus weisen typische pBuM189-Sequenzen im Durchschnitt 1,7 % wiederholungs-spezifische Nukleotid-Substitutionen auf, während im Juan/4-Wiederholung 30 gefunden wurden (z. B. T an den Positionen 11 und 42). Ein so hoher Grad an Nukleotiddivergenz deutet darauf hin, dass die Juan/4-Wiederholung zu einer anderen pBuM189-Subfamilie gehören könnte. Aus theoretischer Sicht hängt der Ursprung von satDNA-Subfamilien von Variablen wie der Kopienzahl, der genomischen Verteilung der satDNA-Familie und den relativen Raten nicht-reziproker Austausche innerhalb und zwischen nicht-homologen Chromosomen ab (DOVER 1986).
Wenn das Juan/4-Wiederholungsmuster aus der Probe ausgeschlossen wird, sinkt die durchschnittliche Nukleotidvariabilität der verbleibenden 62 pBuM189-Sequenzen auf 3,7 %. Dieser Wert ist fast identisch mit den 3,8 %, die von KUHN et al. (1999) basierend auf fünf pBuM189-Sequenzen berechnet wurden, die nur aus einer D. buzzatii-Population (Fundort 5) isoliert wurden.
Unter den anderen pBuM189-Wiederholungen ist Ticu/4 aus dem Fundort (14) am ähnlichsten zu Juan/4, da es zwei spezifische Nukleotide (G an Position 70 und A an Position 88) und ein Indel (Position 49) aufweist, die ansonsten exklusiv für die Juan/4-Wiederholung sind, sowie mehrere andere Substitutionen, die sowohl bei Juan/4 als auch bei anderen Wiederholungen gemeinsam sind (z. B. T an Position 36 oder A an Position 133). Die Ticu/4-Wiederholung selbst ist die am stärksten divergente pBuM189-Wiederholung (im Durchschnitt 10,2 %) nach Juan/4 und weist 6 wiederholungsspezifische Nukleotidsubstitutionen (Positionen 1, 69, 79, 97, 98, 167 und 188) auf. Daher muss Ticu/4 als ein intermediärer evolutionärer Schritt zwischen „typischen“ pBuM189-Wiederholungen und der stark divergierten Juan/4-Wiederholung betrachtet werden. Interessanterweise stammen sowohl die seltenen Wiederholungen Juan/4 als auch Ticu/4 aus Argentinien, wo die Art wahrscheinlich ihren Ursprung hat (WASSERMAN 1962; CARSON und WASSERMAN 1965).
Nach theoretischen und empirischen Daten sollte die Differenzierung von satDNA-Sequenzen gemäß dem Konzept der konzertierten Evolution (DOVER 1986; ELDER und TURNER 1994) entstehen. Mit anderen Worten, satDNA-Arrays aus verschiedenen Populationen oder Arten könnten durch den Effekt molekularer Mechanismen wie ungleiche Crossing-over und Genkonversion, gekoppelt mit genetischer Isolation und genetischem Drift, für verschiedene Mutationen homogenisiert werden.
Die Nukleotid-Ausrichtung der 63 pBuM189-Wiederholungen aus 14 D. buzzatii-Populationen (im Durchschnitt 4,5 % Wiederholungen pro Population) zeigte keine spezifische Nukleotidersetzung oder Indel, die eine spezifische Population oder Gruppen geografisch naher Populationen unterscheiden könnte (Abb. 2). In einigen Fällen teilten mehr als zwei Klone identische Nukleotidsequenzen, wie im Fall der Klone Capao/3, Grao/3, M.Cha/6 und Catam/1 oder M.Cha/1 und Santi/4, aber sie stammen aus ziemlich weit entfernten geografischen Lokalitäten. Andererseits wurden in einer einzigen Population keine identischen Wiederholungen gefunden.
Die genetischen Distanzen zwischen den 63 pBuM189 Nukleotidsequenzen wurden nach der Methode von Kimura mit zwei Parametern (KIMURA 1980) berechnet und ein „Neighbor Joining“-Dendrogramm (SAITOU und NEI 1987) wurde erstellt (Abb. 3). Die Juan/4-Sequenz befand sich in einem deutlich separaten Zweig von den anderen pBuM189-Wiederholungen, was die Annahme unterstützt, dass sie zu einer anderen pBuM189 satDNA-Unterfamilie gehört. Die Ticu/4-Wiederholung befand sich in einem Zweig zwischen der Juan/4- und den verbleibenden 61 pBuM189-Wiederholungen. Innerhalb der verbleibenden 61 pBuM189-Wiederholungen konnte kein Cluster beobachtet werden, das pBuM189-Sequenzen aus einer Population oder Gruppen geografisch naher Populationen enthielt.
Insgesamt deuten die Daten auf hohe Ähnlichkeiten in der pBuM189-Sequenz unter D. buzzatii-Populationen hin, die in einigen Fällen mehr als 3000 km voneinander entfernt sind. Ein solches Fehlen von interpopulationaler Differenzierung von satDNA steht im Einklang mit mtDNA-Daten, die einen hohen Genfluss und sehr wenig Populationsstruktur in den meisten Verbreitungsgebieten von D. buzzatii in Südamerika anzeigen (ROSSI et al. 1996; DE BRITO et al. 2002). Dementsprechend muss es eine adaptive Erklärung für den Verlust des chromosomalen Inversionspolymorphismus in brasilianischen Populationen (im Vergleich zu argentinischen Populationen) geben, wie von FIGUEIREDO und SENE (1992) berichtet.
Derzeit sind der Chaco (nördliches Argentinien und Ost-Bolivien) und die Caatinga (Nordost-Brasilien) die einzigen beiden ausgedehnten Gebiete mit arider Vegetation in Südamerika, die eine hohe Dichte und Vielfalt an Kaktusarten aufweisen. In anderen Gebieten sind Kaktusse weniger häufig und unregelmäßig verteilt. Das Gleiche gilt für D. buzzatii-Populationen, die auf Kaktusse zum Fressen und Fortpflanzen angewiesen sind. Es wird allgemein akzeptiert, dass das globale Klima während des Pleistozäns wiederkehrenden Zyklen von kaltem-trockenem und heißem-feuchtem Wetter unterlag, als mindestens vier große Gletscherereignisse und viele kleinere stattfanden. Zum Beispiel war das Klima während des letzten Gletscherereignisses (vor 18.000 bis 13.000 Jahren) in Südamerika trocken, und der Chaco war durch xerophytische Formationen im zentralen Brasilien und an der Atlantikküste mit den Caatingas verbunden (AB'SABER 1977). Folglich hatte D. buzzatii wahrscheinlich eine viel kontinuierlichere Verbreitung als das, was heute beobachtet wird. Eine solche Situation könnte den Genfluss unter den südamerikanischen D. buzzatii-Populationen in großem Maße ermöglicht haben.
Bis heute wurden die meisten in der Literatur beschriebenen Satelliten-DNAs von Individuen einer einzigen Linie oder Lokalität isoliert. Die vorliegenden Daten sind eines der wenigen Beispiele, die Informationen über die Variabilität der satDNA-Sequenzen in natürlichen Populationen einer weit verbreiteten Art liefern.
Autoren: Gustavo C. S. Kuhn, Fernando F. Franco, Wilson A. Silva Jr, Nilce M. Martinez-Rossi und Fabio M. Sene
Referenzen:
- Ab'Saber, N. A. 1977. Espac¸os ocupados pela expansão dos climas secos da América do Sul, por ocasião dos períodos glaciais quaternários. – Paleoclimas 3: 1 – 19.
- Bachmann, L., Raab, M. und Sperlich, D. 1989. Satelliten-DNA und Speziation: eine artspezifische Satelliten-DNA von Drosophila guanche. – Z. Zool. Syst. Evol. Forsch. 27: 84 – 93.
- Baimai, V., Sene, F. M. und Pereira, M. A. Q. R. 1983. Heterochromatin und karyotypische Differenzierung einiger neotropischer Kaktusbrutarten der Drosophila repleta-Gruppe. – Genetica 67: 81 – 92.
- Barker, J. S. F., Sene, F. M., East, P. P. et al. 1985. Allozyme- und chromosomale Polymorphismus von Drosophila buzzatii in Brasilien und Argentinien. – Genetica 67: 161 – 170.
- Carson, H. L. und Wasserman, M. 1965. Ein weit verbreiteter chromosomaler Polymorphismus in einer weit verbreiteten Art, Drosophila buzzatii. – Am. Nat. 99: 111 – 115.
- De Brito, R. A., Manfrin, M. H. und Sene, F. M. 2002. Mitochondriale DNA-Phylogeographie brasilianischer Populationen von Drosophila buzzatii. – Genet. Mol. Biol. 2: 161 – 171.
- Dover, G. A. 1986. Molekularer Antrieb in Multigenfamilien: wie biologische Neuheiten entstehen, sich verbreiten und assimiliert werden. – Trends Genet. 2: 159 – 165.
- Elder, F. J. und Turner, B. J. 1994. Konzertierte Evolution auf Populationsebene: Pupfish HindIII Satelliten-DNA-Sequenzen. – Proc. Natl Acad. Sci. 91: 994 – 998.
- Figueiredo, V. L. C. und Sene, F. M. 1992. Chromosomenvariabilität in brasilianischen Populationen von Drosophila buzzatii (Diptera, Drosophilidae). – Revta. Bras. Biol. 52: 555 – 561.
- Fontdevila, A., Ruiz, A., Ocana, J. et al. 1982. Die evolutionäre Geschichte von Drosophila buzzatii. II. Wie sehr hat sich der chromosomale Polymorphismus bei der Kolonisierung verändert? – Evolution 36: 843 – 851.
- Frydenberg, J., Pertoldi, C., Dahlgaard, J. et al. 2002. Genetische Variation in ursprünglichen und kolonialisierenden Drosophila buzzatii-Populationen, analysiert durch Mikrosatelliten-Loci, die mit einer neuen PCR-Screening-Methode isoliert wurden. – Mol. Ecol. 11: 181 – 190.
- Halliburton, R. und Barker, J. S. F. 1993. Mangel an mitochondrialer DNA-Variation in australischen Drosophila buzzatii. – Mol. Biol. Evol. 10: 484 – 487.
- Kimura, M. 1980. Eine einzige Methode zur Schätzung der evolutiven Rate von Basenaustauschen durch vergleichende Studien von Nukleotidsequenzen. – J. Mol. Evol. 16: 111 – 120.
- King, L. M. und Cummings, M. P. 1997. Die Variation der Satelliten-DNA-Wiederholungssequenzen ist niedrig in drei Arten der Aaskäfer der Gattung Nicrophorus (Coleoptera: Silphidae). – Mol. Biol. Evol. 14: 1088 – 1095.
- Kuhn, G. C. S., Bollgönn, S., Sperlich, D. et al. 1999. Charakterisierung einer artspezifischen Satelliten-DNA von Drosophila buzzatii. – J. Zool. Syst. Evol. Res. 37: 109 – 112.
- Kumar, S., Tamura, K., Jakobsen, I. B. et al. 2001. MEGA2: Software zur Analyse molekularer evolutionärer Genetik. – Arizona State Univ.
- Pereira, M. A. Q. R., Vilela, C. R. und Sene, F. M. 1983. Anmerkungen zu Brut- und Futterstätten einiger Arten der repleta-Gruppe der Gattung Drosophila (Diptera, Drosophilidae). – Ciência e Cultura 35: 1313 – 1319.
- Rossi, M.S., Barrio, E., Latorre, A. et al. 1996. Die evolutionäre Geschichte von Drosophila buzzatii. XXX. Mitochondrialer DNA-Polymorphismus in ursprünglichen und kolonialisierenden Populationen. – Mol. Biol. Evol. 13: 314 – 323.
- Ruiz, A., Cansian, A. M., Kuhn, G. C. S. et al. 2000. Das Drosophila serido-Speziesrätsel: neue Puzzlestücke zusammenfügen. – Genetica 108: 217 – 227.
- Saitou, N. und Nei, M. 1987. Die Nachbarverknüpfungsmethode: eine neue Methode zur Rekonstruktion phylogenetischer Bäume. – Mol. Biol. Evol. 4: 406 – 425.
- Tidon-Sklorz, R. und Sene, F. M. 1995. Fauna von Drosophila (Diptera, Drosophilidae) im nördlichen Gebiet der „Cadeia do Espinhac¸o'', Bundesstaat Minas Gerais und Bahia, Brasilien: biogeografische und ökologische Aspekte. – Iheringia Ser. Zool. 78: 85 – 94.
- Tidon-Sklorz, R., Vilela, C. R., Sene, F. M. et al. 1994. Die Gattung Drosophila (Diptera, Drosophilidae) in der Serra do Cipó, Bundesstaat Minas Gerais, Brasilien. – Revta. Bras. Ent. 38: 627 – 637.
- Vilela, C. R., Sene, F. M. und Pereira, M. A. Q. R. 1980. Zur Drosophila-Fauna von Chaco und den östlichen Hängen der Anden in Argentinien. – Revta. Bras. Biol. 40: 837 – 841. Vilela, C. R., Pereira, M. A. Q. R. und Sene, F. M. 1983. Vorläufige Daten zur geografischen Verbreitung von Drosophila-Arten innerhalb morphoklimatischer Domänen Brasiliens. II. Die repleta-Gruppe. – Ciên. Cult. São Paulo 35: 66 – 70.
- Wasserman, M. 1962. Zytologische Studien der repleta-Gruppe der Gattung Drosophila. V. Die mulleri-Untergruppe. – Univ. Texas Publ. 6205: 85 – 118.
- Wu, W. L., Wang, J. P., Tseng, M. C. et al. 1999. Klonierung und genetische Variabilität einer HindIII repetitiven DNA in Acrossocheilus paradoxus (Cyprinidae). – Genome 42: 780 – 788.